Coordinates¶
Primary and Secondary Coordinates are manually curated and verified representative genomic coordinates of the variant (Chromosome, Start, Stop, Reference base, and Variant base) for the assigned reference assembly (e.g., GRCh37).
Understanding Coordinates¶
Primary Coordinates are generated for all Variants. Secondary Coordinates are utilized for structural variants involving two loci (e.g., fusion variants).
Curating Coordinates¶
Choosing representative coordinates
Although multiple genomic changes can often lead to functionally equivalent alterations (e.g., same amino acid change), CIViC uses representative coordinates to provide user-friendly variant context rather than enumerate all possible alterations that could cause the variant. When choosing a representative variant, it is preferable to use the most common or highly recurrent alteration observed. Genomic coordinates are 1-based with left-shifted normalization and include a specified reference assembly (GRCh37 preferred). Based on manually curated representative coordinates, an automated linkout to the ClinGen Allele Registry is created. This link provides additional information such as unique and referenceable identifiers for every registered variant with links to additional resources (e.g., gnomAD, ClinVar). The ClinGen Allele Registry also provides coordinate conversions between different builds of the human references genome and alternative cDNA transcript sequences. If the required ClinGen Allele does not yet exist, the curator should create a ClinGen account and register it.
Choosing a representative transcript
Multiple transcripts can often be expressed for a single gene. For this reason, a specific protein coding alteration, resulting from a genomic change, should always be expressed relative to a specific/individual transcript sequence. CIViC representative transcripts use the Ensembl archived version 75 (GRCh37), and should always include the transcript version number (i.e., ENST00000078429.1 instead of ENST00000078429). There is rarely only one correct transcript. Representative transcripts must contain the variant but are otherwise chosen based on priority criteria such as: wide use in the literature, having the longest open reading frame or most exons, containing the most common exons between transcripts, or having the widest genomic coordinates. These are consistent with Ensembl’s glossary definition of canonical.
Wherever possible curators should use the Ensembl MANE Select Transcript.
Curation Practice for SNVs and Small Indels¶
Variant annotation for single nucleotide variants (SNV) and small insertion/deletions (indels) follow a 1-based coordinate system and utilize left-shifted normalization. Reference positions are indicated in green and variant positions in blue. Below in Figure 1, each representation is the text that would be entered into the CIViC Reference Base and Variant Base fields in the Variant Suggested Revision form.

When selecting a representative variant, utilize the most specific and recurrent variant, whenever possible. For example, there are more than 70 insertions listed in COSMIC that lead to the highly recurrent NPM1 W288fs mutation; however, one 4bp insertion (also known as NPM1-A) accounts for more than 90% of all variant entries. Therefore, the coordinates associated with the NPM1-A variant were chosen as the representative coordinates for the NPM1 W288fs variant in CIViC.
For complex variants such an DelIns variants (combination of both deleted and inserted bases) you should follow the logic of the HGVS nomenclature. Specifically, use the start/stop coordinates to indicate the reference genome position of bases deleted as you would for a small deletion. Then, under Reference Base(s) show the corresponding sequence of bases deleted. Finally, under Variant Base(s) show the sequence of bases inserted (e.g. NC_000003.11:g.10183874_10183881delinsCG would be Chr: 3, Start: 10183874, Stop: 10183881, Ref Bases: ACGGGCCC, Var Bases: CG).
For complex variants such as SNVs in genes involved in fusions (e.g., EML4-ALK C1156Y), enter the genomic position of the SNV.
Categorical variants involving a single amino acid can be indicated by using the three or two base pairs of the corresponding triplet codon that could result in a SNV at that site (see BRAF V600 for example).
Curation Practice for Categorical and Large-scale Variants¶
Variant annotations for large-scale rearrangements or categorical variants aim to utilize the minimum genomic space that encompasses the range of variants observed for that gene. For example, although PIK3CA amplification can encompass much larger genomic coordinates, the outermost coordinates of the gene PIK3CA are used to define the start and stop coordinates. Similarly, categorical variants that contain mutations within the same domain or exon use the outermost coordinates of that domain or exon. Fusion coordinates are based on the closest exon boundary included in the fusion. Although multiple breakpoints can occur, the most common breakpoint is prefered for the representative coordinate.
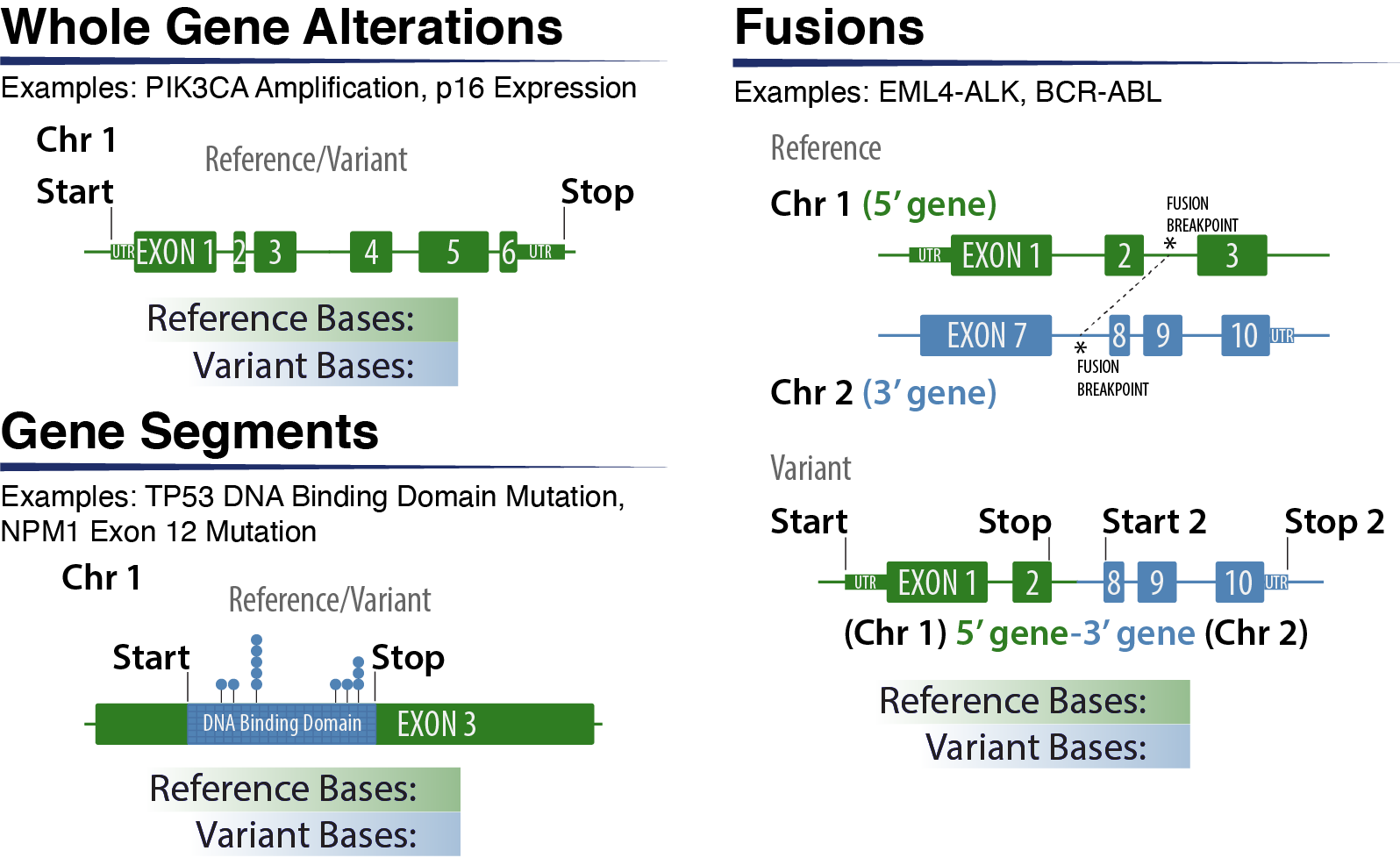
Use coordinates that would encompass the most variants that fit the Variant Name to aid others using coordinates to find relevant and similar variants.
For fusions:
Names are always written in a 5’ to 3’ order (e.g. 5’ BCR-ABL 3’);
The Variant is placed under the most ‘important’ gene - e.g. kinase domain - (not repeated under both) which is often the 3’ gene;
Coordinates represent the entire putative fusion transcript including start to end of fused/involved exons of 5’ transcript partner (primary coordinates) and start to end of fused/involved exons of 3’ transcript partner (secondary coordinates).
Curation Practice for representative transcripts¶
Genes often have multiple transcript representations. CIViC utilizes Ensembl v75 for transcript annotations. The representative transcript for WT1 depicted in blue below was chosen because it has the widest outer coordinates with the most common exons compared to the other transcripts depicted in green. This transcript is further highlighted by *** because it is also designated as the “canonical transcript” by Ensembl using select criteria defined in their glossary of terms.

There is no one ‘right’ answer for representative transcript.
It must:
contain the variant (except in rare cases like promoter mutations);
be based on an Ensembl transcript and include the transcript version.
It may:
be the transcript with the longest ORF or most exons;
be the transcript that contains the ‘canonical exons’ that are used in many transcripts;
be the variant that has the greatest outer coordinates;
be the transcript that is widely used in literature;
be a transcript that is compatible with interpretation/visualization in the primary literature source.
An IGV reference transcript file containing Ensembl (v75) transcripts can be obtained here (Ensembl-v75_build37-hg19_UcscGenePred_CIViC-Genes.ensGene).
Ensembl canonical transcripts are designated by ***.
Selection of Representative Transcripts for intronic or regulatory variants follow a similar pattern as protein coding variants.